Training the next generation of talented pediatric research coordinators is an essential part of CHOC Research’s mission to support top-quality research that improves children’s lives and well-being. Chief Scientific Officer Dr. Terence Sanger says.

CHOC Pediatrica
News and Information for Pediatric Healthcare Professionals
Training the next generation of talented pediatric research coordinators is an essential part of CHOC Research’s mission to support top-quality research that improves children’s lives and well-being. Chief Scientific Officer Dr. Terence Sanger says.
CHOC is one of the top nine centers in the country doing cutting-edge cerebellum research.

The annual conference signals a new era under the Rady Children’s Health umbrella.
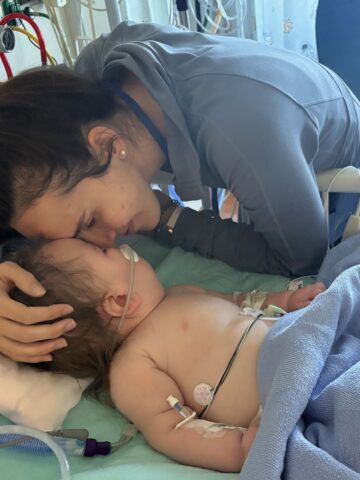
With time not on their side, CHOC care team pulls out all stops to save life of Emery Kline.

Panel discussions also were held on precision medicine, fairness in AI, precision management of psychiatric conditions, and other issues.
The new study is an example of how CHOC and UCLA Health are transforming the way cardiac care is being delivered in Southern California.